

Abstract
Background: B-lineage acute lymphoblastic leukemia (B-ALL) is a highly heterogeneous disease that contains different biological and prognostic features. Relapse and drug resistance are the critical clinical issues of B-ALL. To improve outcome and potentially reduce side effects and long-term sequelae from current therapy, the development of novel, biologically relevant, and molecular target therapies is critical. Previous studies have shown that toll-like receptor 9 (TLR9) is expressed at almost all stages of B cell development. Targeting TLR9 expressed B-cell malignancies with TLR9 agonists, CpG oligodeoxynucleotides, can have multiple antitumor effects. However, due to the heterogeneous on B-cell malignancies, the impact of TLR9 agonist on different subtypes of B-ALL remains unknown. This study is to determine the antitumor effects and mechanism of a TLR9 agonist, CpG 685, for treating Ph(+) vs Ph(-) B-ALL subtypes.
Methods: Real-time quantitative PCR was used to determine the expression level of TLR9 in different subtypes of clinical B-ALL samples. WST-1 cell viability assay and flow cytometry were used to detect the effects of CpG 685 on proliferation, apoptosis, cell cycle, and the regulation of costimulatory molecules such as CD40 and CD80 expression on Ph(+) vs Ph(-) B-ALL cell lines. Western blot was used to detect the activation of TLR9 downstream molecules by CpG 685 stimulations. Vital molecular inhibitors were used to clarify the signaling pathway of CpG 685-induced apoptosis in B-ALLs. The relationship between CpG 685-treated and no CpG 685 group analyzed by SPSS 24.0 software (SPSS, Chicago, IL), p<0.05 was considered meaningful.
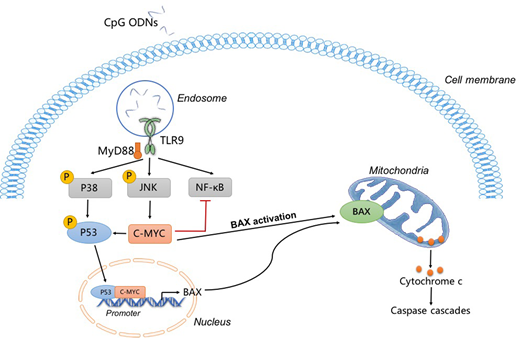
Results: The expression of TLR9 was found in more than 95% of patients with B-ALL (n=49). Treatment with CpG 685 upregulated the expression of surface molecules on B-ALL cells, especially CD40 and MHC-Ⅰ. CpG 685 effectively inhibited proliferation, induced apoptosis and G1-phase arrest in Ph(-) B-ALL cell line during a 3-day culture (p<0.01). CpG 685-induced apoptosis on BLIN-1 cell line was dependent on the high expression of C-MYC and the activation of the P38/P53/BAX signaling pathway. The activation of C-MYC can upregulate the expression of P53 via the ARF/Mdm-2 pathway, and further promote the activation of BAX, releasing mitochondria to release cytochrome C, thereby promoting apoptosis. The previous study has shown that CpG ODNs induce proliferation in mouse primary B cells through sustained increases in NF-κB activation due to the low expression level of C-MYC. However, in our study, because of the high expression levels of C-MYC, CpG ODNs trigger a transient NF-κB activation in B-ALL, leading to the occurrence of apoptosis eventually. Interestingly, the mechanism of Ph(+) B-ALL resistance to CpG 685 was that BCR-ABL inhibits activation of BAX through multiple pathways. By using imatinib to block BCR-ABL kinase, CpG 685 is useful in anti-leukemia effect on Ph(+) B-ALL cells due to the re-activation of BAX. Besides, Sup-B15 was an imatinib resistance cell line, the combinational use of CpG 685 and imatinib to treat Ph(+) B-ALL cells can also reverse the drug resistance of imatinib treatment alone.
Conclusion: This study demonstrated that TLR9 is a potential novel target for B-ALL therapy. CpG 685 has multiple anti-tumor effects through direct killing of B-ALL cells and enhancing immunogenicity of B-ALL cells. For CpG 685 monotherapy, screening for B-ALL patients with the C-MYC high expression levels and without Philadelphia chromosome are the key biomarkers in the treatment. CpG 685 can also reverse imatinib resistance in patients with B-ALL and may serve as a potential useful anti-leukemia agent for combinational therapy of Ph(+) B-ALL.
No relevant conflicts of interest to declare.

